
Svi iLive sadržaji medicinski se pregledavaju ili provjeravaju kako bi se osigurala što je moguće točnija činjenica.
Imamo stroge smjernice za pronalaženje izvora i samo povezujemo s uglednim medijskim stranicama, akademskim istraživačkim institucijama i, kad god je to moguće, medicinski pregledanim studijama. Imajte na umu da su brojevi u zagradama ([1], [2], itd.) Poveznice koje se mogu kliknuti na ove studije.
Ako smatrate da je bilo koji od naših sadržaja netočan, zastario ili na neki drugi način upitan, odaberite ga i pritisnite Ctrl + Enter.
Lisencefalija mozga
Medicinski stručnjak članka

Posljednji pregledao: 12.07.2025
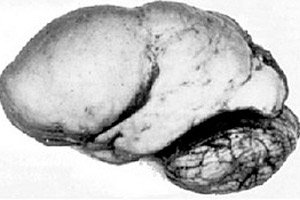
Među organskim cerebralnim patologijama ističe se kongenitalna anomalija razvoja mozga kao što je lisencefalija, čija je bit gotovo glatka površina korteksa njegovih hemisfera - s nedovoljnim brojem vijuga i brazda. [ 1 ]
U potpunom nedostatku konvolucija definira se agirija, a prisutnost nekoliko širokih ravnih konvolucija naziva se pahigirija. Ovi defekti, kao i neke druge redukcijske deformacije mozga, imaju kod Q04.3 u ICD-10.
Epidemiologija
Prema statistikama o rijetkim bolestima, postoji 1-1,2 slučaja lisencefalije na 100 tisuća novorođenčadi. [ 2 ], [ 3 ]
Prema nekim podacima, do 25-30% slučajeva klasične lisencefalije opaža se kod djece s Miller-Diekerovim sindromom; točkaste mutacije i delecije gena LIS1 i DCX otkrivaju se kod gotovo 85% pacijenata. [ 4 ]
Genetske studije 17 gena povezanih s lisencefalijom pokazale su da mutacija ili delecija LIS1 odgovorna je za 40% pacijenata, a 23% ih je povezano s mutacijom DCX, a slijede TUBA1A (5%) i DYNC1H1 (3%).[ 5 ]
Uzroci lisencefalija
Svi poznati uzroci formiranja moždane kore (cortex cerebri) gotovo ili potpuno bez vijuga i brazda, koji povećavaju "radno područje" ljudskog mozga i osiguravaju "produktivnost" središnjeg živčanog sustava, povezani su s poremećajima u njegovom perinatalnom razvoju. To jest, lisencefalija se razvija kod fetusa. [ 6 ]
Neuspjeh u formiranju slojeva moždane kore fetusa kod lisencefalije rezultat je abnormalne migracije neurona koji je tvore ili preranog prestanka tog procesa.
Ovaj proces, koji je bitan za cerebrokortikalnu histogenezu, odvija se u nekoliko faza od 7. do 18. tjedna trudnoće. A s obzirom na povećanu osjetljivost na genetske mutacije, kao i na razne negativne fizičke, kemijske i biološke utjecaje, svako odstupanje od norme može dovesti do netočne lokalizacije neurona s mogućim stvaranjem zadebljanog sloja sive tvari korteksa bez karakteristične strukture. [ 7 ]
U nekim slučajevima, lisencefalija kod djece povezana je s Miller-Diekerovim, Walker-Warburgovim ili Norman-Robertsovim sindromima.
Pročitajte i – Razvojni defekti mozga
Faktori rizika
Osim mutacija u nekim genima, faktori rizika za rođenje djeteta s tako teškim defektom uključuju gladovanje kisikom (hipoksiju) fetusa; nedovoljnu opskrbu mozga krvlju (hipoperfuziju); akutni cerebrovaskularni inzult u obliku perinatalnog moždanog udara; patologije posteljice; virusne infekcije trudnice (uključujući TORCH); [ 8 ] probleme s općim metabolizmom i funkcijom štitnjače; pušenje, alkohol, psihotropne i narkotičke tvari; upotrebu niza lijekova; povećanu razinu zračenja. [ 9 ]
Patogeneza
Nisu svi slučajevi lisencefalije uzrokovani patogenezom kromosomskih abnormalnosti i genskih mutacija. No, poznati su neki geni koji kodiraju proteine koji igraju važnu ulogu u ispravnom kretanju neuroblasta i neurona duž radijalnih glijskih stanica - kako bi formirali moždanu koru. A mutacije tih gena dovode do ove patologije. [ 10 ]
Posebno se radi o sporadičnim mutacijama (bez nasljednosti) gena LIS1 na kromosomu 17, koji regulira citoplazmatski motorni protein mikrotubula dinein, kao i gena DCX na X kromosomu, koji kodira protein doublecortin (lisencefalin-X). [ 11 ]. U prvom slučaju, stručnjaci definiraju klasičnu lisencefaliju (tip I), u drugom - X-vezanu. [ 12 ]
Kada se gen FLN1, koji kodira fosfoprotein filamin 1, izbriše, proces usmjerene neuronske migracije možda uopće neće započeti, što dovodi do potpunog nedostatka konvolucija (agirije). [ 13 ]
Mutacije su identificirane u genu CDK5, koji kodira enzim kinazu – katalizator za unutarstanični metabolizam, regulirajući stanični ciklus u neuronima središnjeg živčanog sustava i osiguravajući njihovu normalnu migraciju tijekom prenatalnog formiranja moždanih struktura.
Abnormalne promjene u genu RELN na kromosomu 7 koje uzrokuju kortikalne giralne defekte kod Norman-Robertsovog sindroma rezultiraju nedostatkom izvanstaničnog glikoproteina reelina, koji je potreban za regulaciju migracije i pozicioniranja živčanih matičnih stanica tijekom razvoja korteksa cerebri.[ 14 ], [ 15 ], [ 16 ]
Gen ARX kodira nearistalens homeobox protein, transkripcijski faktor koji igra važnu ulogu u prednjem mozgu i drugim tkivima.[ 17 ] Djeca s ARX mutacijom imaju druge simptome, poput nedostajućih dijelova mozga (ageneza corpus callosuma), abnormalnih genitalija i teške epilepsije.[ 18 ],[ 19 ]
Nekoliko gena povezano je s lisencefalijom. Ti geni uključuju VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A i CDK5.[ 20 ]
Citomegalovirus (CMV) povezan je s razvojem lisencefalije zbog smanjene opskrbe krvlju fetalnog mozga. Ozbiljnost CMV infekcije ovisi o gestacijskoj dobi. Rana infekcija vjerojatnije će uzrokovati lisencefaliju jer se migracija neurona događa rano u trudnoći.[ 21 ]
Osim toga, mehanizam nastanka ove anomalije uključuje nepotpuni ili kasniji prestanak migracije neurona iz periventrikularne generativne zone u moždanu koru. U takvim slučajevima razvija se ili nepotpuna lisencefalija ili pahigirija, u kojoj se formira nekoliko širokih žljebova i vijuga (ali većina ih je odsutna).
Simptomi lisencefalija
Prvi znakovi ove patologije (u odsutnosti prethodno spomenutih sindroma) možda se neće pojaviti odmah nakon rođenja, već nakon mjesec i pol do dva. Najčešće se opažaju sljedeći klinički simptomi lisencefalije:
- mišićna hipotonija, često u kombinaciji sa spastičnom paralizom;
- konvulzije i generalizirani toničko-klonički napadaji (u obliku opistotonusa);
- teška mentalna retardacija i zaostajanje u rastu;
- oštećenje neuroloških i motoričkih funkcija.
Problemi s gutanjem uzrokuju poteškoće u hranjenju bebe. [ 22 ]
Visok stupanj neuromotornog oštećenja često se manifestira kao tetraplegija - paraliza svih udova. Moguća je deformacija šaka, prstiju na rukama ili nogama.
Kod Norman-Robertsovog sindroma s lisencefalijom tipa I uočavaju se kraniofacijalne anomalije: teška mikrocefalija, nizak nagib čela i izbočen široki nosni most, široko postavljene oči (hiperterlorizam), nerazvijenost čeljusti (mikrognatija). [ 23 ]
Miller-Diekerov sindrom može se također okarakterizirati abnormalno malom veličinom glave sa širokim, visokim čelom i kratkim nosom, udubljenjima u sljepoočnicama (bitemporalnim udubljenjima) i nisko postavljenim, deformiranim ušima.
Teški sindrom lisencefalije karakterizira mikrocefalija, smanjenje veličine očnih jabučica (mikroftalmija), u kombinaciji s retinalnom displazijom, opstruktivnim hidrocefalusom i odsutnim ili hipoplastičnim corpus callosumom.
Komplikacije i posljedice
Među komplikacijama ove anomalije, stručnjaci navode kršenje funkcije gutanja (disfagija) i gastroezofagealni refluks; refraktornu (nekontroliranu) epilepsiju; česte infekcije gornjih dišnih putova; upalu pluća (uključujući kroničnu aspiraciju).
Dojenčad s lisencefalijom može imati kongenitalne organske srčane probleme u obliku defekta atrijskog septuma ili složene srčane mane s cijanozom (tetralogija Fallot). [ 24 ]
Posljedice postnatalnog zaostajanja u rastu u većini slučajeva rezultiraju smrću unutar 24 mjeseca nakon rođenja.
Dijagnostika lisencefalija
Dijagnoza započinje fizičkim pregledom djeteta, proučavanjem medicinske anamneze roditelja te povijesti trudnoće i poroda.
Tijekom trudnoće može biti potrebno testiranje fetalne DNK bez stanica, amniocenteza ili uzimanje uzoraka korionskih resica. [ 25 ] Za više informacija pogledajte – Prenatalna dijagnostika kongenitalnih bolesti
Instrumentalna dijagnostika koristi se za vizualizaciju moždanih struktura i procjenu njihovih funkcija:
- kompjuterizirana tomografija mozga;
- magnetska rezonancija (MRI) mozga;
- elektroencefalogram (EEG). [ 26 ]
Tijekom trudnoće, lisencefalija na ultrazvuku fetusa nakon 20-21 tjedna može se posumnjati u odsutnosti parijeto-okcipitalnih i kalkarinskih žljebova te anomalije Sylvijske fisure mozga.
Diferencijalna dijagnoza
Provodi se diferencijalna dijagnostika s drugim sindromima kongenitalnih cerebralnih mana.
Postoji više od 20 vrsta lisencefalije, od kojih se većina svrstava u 2 glavne kategorije: klasična lisencefalija (tip 1) i lisencefalija tipa "cobblestone" (tip 2). Svaka kategorija ima slične kliničke manifestacije, ali različite genetske mutacije.[ 27 ]
Pregled mozga kod lisencefalije tipa I pokazuje moždanu koru s četiri sloja umjesto šest koji se vide kod normalnih pacijenata, dok je kod lisencefalije tipa 2 moždana kora neorganizirana i izgleda grudasto ili nodularno zbog potpunog pomicanja moždane kore nakupinama kortikalnih neurona odvojenih gliomezenhimskim tkivom. Pacijenti su također imali abnormalnosti mišića i očiju.
- Klasična lisencefalija (tip 1):
- LIS1: Izolirana lisencefalija i Miller-Diekerov sindrom (lisencefalija povezana s facijalnim dismorfizmom). [ 28 ]
- LISX1: mutacija gena DCX. U usporedbi s lisencefalijom uzrokovanom mutacijama LIS1, DCX pokazuje korteks sa šest slojeva umjesto četiri.
- Izolirana lisencefalija bez drugih poznatih genetskih defekata
- Kaldrmisane lisencefalije (tip 2):
- Walker-Warburgov sindrom
- Fukuyamin sindrom
- Bolest mišića, očiju i mozga
- Druge vrste ne mogu se svrstati u jednu od dvije gore navedene skupine:
- LIS2: Norman-Robertsov sindrom, sličan lisencefaliji tipa I ili Miller-Diekerovom sindromu, ali bez delecije kromosoma 17.
- LIS3
- LISX2
Mikrolisencefalija: Ovo je kombinacija odsutnosti normalnog kortikalnog nabora i abnormalno male glave. Djeca s pravilnom lisencefalijom imaju normalnu veličinu glave pri rođenju. Djeci s malom veličinom glave pri rođenju obično se dijagnosticira mikrolisencefalija.
Također je važno razlikovati lisencefaliju i polimikrogiriju, koje su različiti defekti u razvoju mozga.
Tko se može obratiti?
Liječenje lisencefalija
Lisencefalija je neizlječiv organski defekt, stoga je moguće samo potporno i simptomatsko liječenje. [ 29 ]
Prije svega, to je uporaba antikonvulziva i antiepileptika, kao i ugradnja gastrostomske cijevi u želudac (ako dijete ne može samostalno gutati). Masaža je korisna.
U slučajevima teškog hidrocefalusa uklanja se cerebrospinalna tekućina.
Prevencija
Stručnjaci preporučuju budućim roditeljima da potraže genetsko savjetovanje, a trudnicama da se pravovremeno registriraju kod opstetričara i ginekologa te obave sve planirane preglede.
Prognoza
Za djecu s lisencefalijom, prognoza ovisi o njezinom stupnju, ali najčešće mentalni razvoj djeteta ne prelazi razinu od četiri do pet mjeseci. I sva djeca s ovom dijagnozom pate od teških psihomotornih poremećaja i teško liječive epilepsije. [ 30 ]
Prema NINDS-u (Američki nacionalni institut za neurološke poremećaje i moždani udar), maksimalni životni vijek za osobe s lisencefalijom je oko 10 godina.